本帖最后由 团子良 于 2023-1-11 10:15 编辑
摘 要 本文主要以FDA,EMA 和NMPA 相关溶出度指导原则为基础,对仿制药溶出度质量标准建立的目的、内涵、一般过程以及特殊考虑进行了论述。提出目前国内研发单位还需进行更加深入的研究,以保证建立更能反映药物内在质量的溶出度质量标准。
药品质量标准是国家对药品质量、规格及检验方法所作的技术规定,是药品生产、供应、使用、检验和药政管理部门共同遵守的法定依据。我国作为一个仿制药大国,在过去很长一个时期,药品质量标准是评价药品安全有效的最重要指标。随着质量源于设计理念的深入人心,药品质量不再是检验出来的,而是设计出来的。但从目前以及未来很长一段时间来看,药品质量标准仍然是监管部门监控药品质量、保障人民群众用药安全的最直接有效的工具之一。但是怎样建立既能辨别药品真伪、纯度,又能反映其内部品质的质量标准,一直是药物研发者和监督者持续研究和共同关注的难题。
口服固体制剂质量标准的内容一般包括: 性状、鉴别、有关物质、溶出度、含量测定等。目前,我国口服固体制剂有关物质的研究和要求基本已和国际接轨,多条溶出曲线对比以降低生物不等效概率的研究也有进行,但涉及在进行了大量溶出度试验研究后,最终如何科学客观地制订溶出度试验质量标准,即通过溶出度质量标准的判断结果就能充分反映出该产品所应具有的内在优良品质、良好体内生物等效性及品质均一性,仍是我国药品研发中较为薄弱的环节。
1、溶出度质量标准制定的一般过程及原则
关于溶出度质量标准的制定,在FDA 1997 年发布的《口服固体速释制剂溶出度试验指导原则》和原国家食品药品监督管理总局于2015 年2月发布的《普通口服固体制剂溶出度试验指导原则》中有部分论述; 但更加系统以及更加详细的论述是在EMA 于2016 年起草、2017 年7 月被采用的《全身作用的口服固体速释制剂仿制药溶出度质量标准的意见书》中。虽然在EMA 发布的指导原则中,仅涉及全身作用的口服固体速释制剂仿制药,但相似的原则同样适用于原研药以及其他产品中。结合上述指导原则以及相关文献,笔者认为溶出度质量标准的制定一般分以下几步。 1.1溶出方法的建立
1.1.1溶出介质的组成及体积
溶出介质一般是基于活性成分的理化性质以及制剂的处方和剂量范围进行选择。通常应采用pH值1.2~6.8的水性介质,一般不应添加表面活性剂,如添加应尽可能少,并需充分验证;体积一般为500,900或1000mL,溶出介质的体积最好能满足漏槽条件。 1.1.2溶出装置 目前被各国监管机构广泛认可的有篮法和桨法以及在此基础上改进并经过验证的方法。一般情况下首选桨法。对于容易产生漂浮的片剂或胶囊,建议采用篮法,当必须采用桨法时,可使用沉降篮或其他适当的沉降装置。 1.1.3转速 一般情况下篮法转速应以100r·min-1起始;桨法的转速为50r·min-1起始。增加转速需要有充分的理由。例如,溶出结果的高变异(≤10min的时间点RSD>20%,后面的时间点RSD>10%),而这种高变异的结果又是由可观察到的流体动力效应或其他因素(如黏片等)造成。然而,增加转速带来的结果就是降低方法的区分力,如果仅仅因为溶出结果的高变异就增加转速是不被允许的,除非根据体内研究结果证明目前条件属于过度区分的情况。但是,不管哪种情况,即使增加转速仍然要保证溶出方法有足够的区分力来控制产品的质量。 1.2溶出方法的区分力
1.2.1选择质控条件
为了将生物等效性试验批的生物等效性研究结果外推至商业批,需要建立有效的溶出度检查方法,该方法能区分不同批次产品的质量,最理想的情况就是所有生物不等效的批次都能被检测发现。 在研发过程中,所有的溶出条件均应与药动学参数进行相关性分析从而找到最合适的溶出条件进行常规检测。但是由于仿制药研究过程中,体内研究数据非常有限,因此要建立能体现出体内外相关性的溶出度检查方法是很困难的,但是在选择溶出条件时,与体内相关性的这个出发点是始终要考虑的。 1.2.2区分力的论证 为了证明溶出度检测方法的适用性,应该采用具有不同质量属性的批次样品进行研究。而这些批次样品的生产需要对某些关键工艺参数或关键物料属性进行有意义的改变。这些改变包括:处方、物料标准和/或工艺参数等,而想使得这些改变更加有意义还需要同时考虑原料药生物药剂学分类(biopharmaceutics classification system,BCS) 及制剂特征。例如: 对于BCS Ⅱ和Ⅳ类药物,原料药的溶解度或溶出特性为体内吸收的限速步骤,那么原料药的粒径分布以及其他一些显著影响药物体内释放属性的改变将可能是有意义的。对于BCS Ⅰ或Ⅲ类药物,胃排空或肠黏膜渗透性可能为体内吸收的限速步骤,那么处方和生产工艺的改变将可能是有意义的,因为处方和工艺的改变可能会影响最终产品的崩解和体外溶出的速率。 通过处方组成的改变来创造一个“劣质批”,但其辅料种类应保持一致,仅仅是部分辅料含量的变化,完全去掉一个或几个特殊的辅料是不被允许的。合适的溶出条件能够将上述改变检出,并用溶出度限度能够区分。 理想情况下,体外溶出试验应该可以预测体内的结果。但在某些情况下不能,因为溶出条件存在过度区分的情况。但是这种溶出条件是被认可的,因为这种情况下只要溶出度不发生改变,体内的等效性是可以保证的。一般情况下,具有不同质量属性产品的体内数据是不容易获得的。因此适合的溶出条件往往是从能够区分不同质量属性产品来确定。 对于BCS 分类Ⅰ和分类Ⅲ的药品,并且在生理pH 范围内具有较高的溶解度且属于快速溶出或非常快速溶出的。此类产品的处方、物料质量标准和/或生产工艺的变化很难从溶出结果中体现。这种情况下,溶出条件不需要更多的验证或者溶出度检查可以采用崩解时限代替。 1.3 溶出度限度的制定
当适当的溶出度试验条件确定以后,下一步要制定合适的溶出度标准。欧洲药典( EP) 的溶出度限度被定义为Q 值,在一个确定的时间点,从能否满足S1,S2 和S3( Ph,Eur.2.9.3) 来区分可接受的批次和不可接受的批次。溶出相似的2 批次样品其平均溶出度结果差异在10% 以内,因此,Q 值推荐设定为生物等效性( bioequivalence,BE) 批次溶出结果减去10%。 一般Q 值被设定在75%~85%,大于85%被认为是没有必要的。一般情况下,取样时间点为15,30和45min,如果采用其他时间点,需进行充分的验证。采用少于15 min 取样时间认为是没有必要的。 溶出度质量标准的区分力是与取样时间点和Q值都紧密关联的: ①BE 批次在15min内溶出≥95%,那么质量标准可以设定在15min Q 值=85%。②BE 批次在15min内溶出≥85%而<95%,那么质量标准的Q 值可以设定为75%,80%或85%,选择最接近BE 批15min溶出结果-10%的那个结果。③BE 批次溶出结果只有在30min后才能≥85%,那么质量标准的Q 值可以设定为75%,80%或85%,选择最接近BE 批次30min溶出结果-10%的那个结果。④BE 批次溶出结果只有在45min后才能≥85% ,那么质量标准的Q 值可以设定为75%,80%或85%,取样时间点在45min。⑤BE 批次在45min后溶出≤85%,那么如果可以设定溶出标准为45min的Q值为75%是可以认可的; 否则需采用两点法进行溶出度控制。溶出度质量标准制定过程可以采用决策树呈现如图1所示。 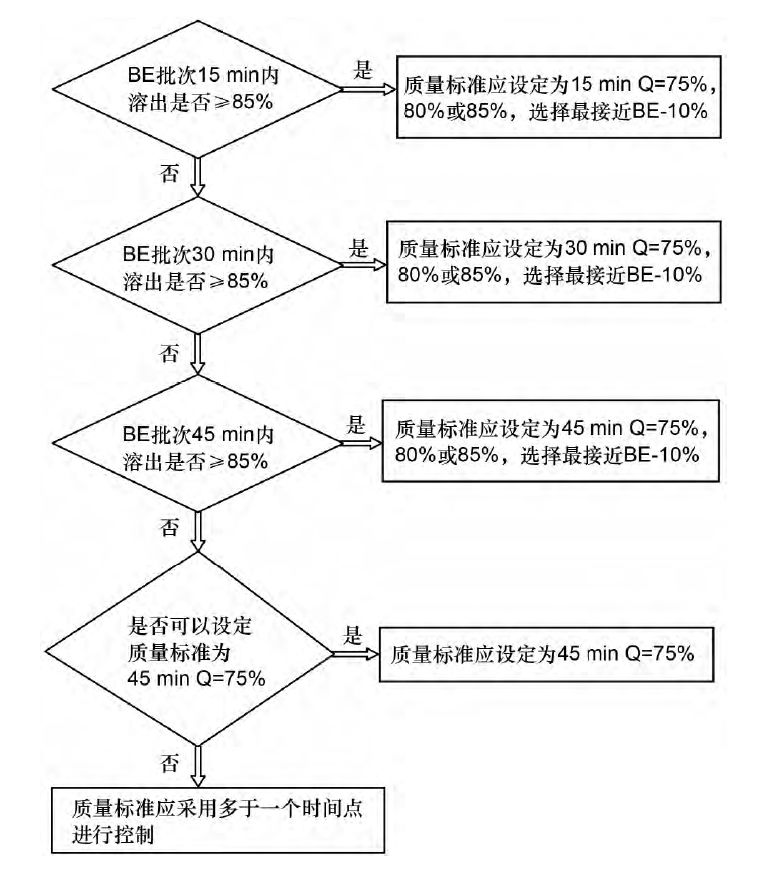
2.溶出度质量标准的验证
口服固体制剂溶出度标准制定的初衷是想反映产品良好的内在品质。最理想的状态是符合溶出度质量标准的产品即具有良好的生物等效性,不符合溶出度质量标准的产品即生物不等效。但这只是一种理想状态,实际中几乎不可能存在这种绝对对应的情况,但是从最理想的情况可以使我们了解到,若想使得溶出度质量标准得到真正的验证就必须通过体内试验。但是基于仿制药有限的体内数据,一般考虑分以下几种情况对溶出度质量标准进行部分验证。 2.1 根据多批次体内行为的样品进行评估
研究过程中有多批次体内试验数据,其中包括生物等效批次的数据和生物不等效批次的数据。从而可以采用这些批次的体内试验结果验证溶出度质量标准的合理性,即生物等效的批次应符合质量标准要求,生物不等效的批次应不符合质量标准。对于仿制药,这种情况实属少数,毕竟多批次体内数据的获取会大大增加仿制药研发的成本。 2.2 边缘批次
采用关键生产工艺变异的极端情况生产出边缘批次,若通过生物等效性试验证明边缘批次与参比制剂等效,那么边缘批次的溶出度可以作为溶出度质量标准的边界,在此溶出度范围内的产品可以认为具有生物等效性。 2.3根据仿制品和参比制剂的体内数据进行评估
评价生物等效性的主要药动学参数为AUC 和Cmax,如果在AUC 基本一致的情况下,Cmax可以部分反映药物体内吸收的快慢。此种情况下,可以根据仿制品和参比制剂Cmax的高低与体外溶出的快慢进行比较,可部分验证溶出度方法的体内预测性。2.4 对于基于BCS 分类豁免生物等效性试验的仿制药由于几乎没有体内数据可以作为参考,所以这种情况下,质量标准限度的制定也不可能采用文中决策树进行确定。那么这种情况该怎么确定产品的溶出度质量标准呢? 在FDA 于2018年8 月发布了《含高溶解性药物的速释口服固体制剂溶出度测试和可接受标准行业指南》中,在不存在可能影响药物吸收的辅料的情况下,在规定的溶出条件下,规定含高溶解性药物的口服固体制剂的可接受标准为30 min 内,Q=80% ,以此作为一个最低的要求。
3.讨论
目前在我国的仿制药研发中,对仿制药与参比制剂多条溶出曲线的对比研究都能做到,但是在进行了大量溶出度试验研究后,最终应如何科学客观地制订溶出度试验质量标准仍是我国仿制药研发过程中的一个薄弱环节。在药物研发过程中,一个药物会涉及许多溶出方法,但定入质量标准的溶出度方法一般仅为一种,因此怎样选择溶出度方法并制定合适的限度要求是需要研究者进行大量论证研究的。因为最终用于上市后产品日常监测的仅是最终制定的溶出度质量标准而非溶出曲线或其他。溶出度质量标准作为反映口服固体制剂内在品质的关键指标,虽然有其局限性,但目前来说,对口服固体制剂在最终反映产品质量、监控工艺稳定性、预测生物等效性方面仍然有着不可替代的作用。 目前国内仿制药研发中,溶出度方法的建立一般分为以下2 种情形,一是原研药的溶出度方法可获得,对于仿制药研发者来说一般会选择原研药的溶出度方法,这不失为一种比较保险的做法。另一种为原研药溶出度的方法不可得或者原研药溶出度方法不适合仿制药时,如何科学、合理的建立溶出度的方法就成为目前的一大难题。本文对仿制药溶出度质量标准建立的一般考虑进行了论述,旨在抛砖引玉,希望可以对仿制药研发过程中溶出度标准的建立提供参考。
| 
